

AUTORES
- Jorge Sánchez Melús. Facultativo Especialista de Área. Servicio de Urgencias. Hospital Ernest Lluch Martín. Carretera de Sagunto-Burgos Km 254, 50300, Calatayud (Zaragoza).
- Victor Pe De La Riva. Residente de 4º Año. Servicio de Urgencias. Hospital Ernest Lluch Martín. Carretera de Sagunto-Burgos Km 254, 50300, Calatayud (Zaragoza).
- Juan Andrés Passarino Iglesias. Residente de 4º Año. Servicio de Urgencias. Hospital Ernest Lluch Martín. Carretera de Sagunto-Burgos Km 254, 50300, Calatayud (Zaragoza).
- Laura Maria Agud Sanz. Enfermera. Servicio de Radiodiagnóstico. Hospital Clínico Universitario «Lozano Blesa». Avda. San Juan Bosco, 15 50009 Zaragoza.
RESUMEN
La poliquistosis renal autosómica dominante es la causa desde el punto de vista genético más frecuente de insuficiencia renal terminal. Es una enfermedad sistémica y progresiva cuya patogenia involucra la formación de quistes renales, aumento del tamaño de órganos extrarrenales (hígado, páncreas, bazo…). No hay diferencia entre razas ni géneros. Puede ser detectada en la infancia, pero las manifestaciones clínicas suelen ocurrir en la tercera década de vida. A continuación, veremos el diagnóstico de este síndrome de forma incidental.
PALABRAS CLAVE
Hematuria, urología, quiste.
ABSTRACT
Autosomal Dominant Polycystic Disease is the most common genetic cause of renal failure worldwide. It is a multisystem and proggresive disease with cyst formation, kidney enlargement and extrarrenal organ involvement (liver, pancreas, spleen…). It occurs in all races. Cysts may be detected in childhood, but clinical manifestations appear between the thrid and fourth decade of life. We will see the diagnose of this syndrome indicentally.
KEY WORDS
Hematuria, urology, cyst.
INTRODUCCIÓN
La poliquistosis renal autosómico dominante (PQRAD) se trata de una enfermedad monogénica multisistémica que se caracteriza por la presencia de múltiples quistes renales bilaterales junto con manifestaciones extrarrenales (quistes en otros órganos, anomalías digestivas, cardiovasculares y musculoesqueléticas)1. Es la enfermedad hereditaria renal más frecuente siendo tercera causa de insuficiencia renal terminal1. Afecta por igual a todas las razas y por igual a ambos sexos. Su herencia como bien vemos es autosómica dominante con penetrancia completa: cada hijo de un padre afectado tiene un 50% de probabilidad de heredar el gen mutado. En su patogenia encontramos dos genes mutados: PKD1 y PKD2. La mayoría de las mutaciones en el PKD1 desarrollan la enfermedad renal terminal a los 50 años de media mientras que la mutación en el PKD2 se manifiesta más tarde (70 años)2. Estos genes codifican la Poliquistina-1 (PC1) y Poliquistina-2 (PC2) respectivamente, ambas asociadas a la membrana. Ambas forman un complejo cuyo papel principal sería la regulación del calcio intracelular3. La disminución de una de las dos por debajo de un nivel crítico lleva consigo un cambio fenotípico que se caracteriza por la incapacidad para mantener la polaridad celular y aumento de proliferación y apoptosis4. Es importante asesorar correctamente al paciente acerca de beneficios y desventajas del diagnóstico de certeza (entre ellas la discriminación en un seguro de enfermedad o en el medio laboral). En pacientes mayores de 18 años el diagnóstico se establece con prueba radiológica, en este caso la ecografía (bajo coste). Otras como la realización de pruebas genéticas están cada día más en auge. Clínicamente se pueden manifestar con gran variedad de síntomas: alteraciones de función renal, hipertensión arterial5 o el dolor (como síntoma más frecuente)6,7.
PRESENTACIÓN DEL CASO CLÍNICO
Varón de 21 años sin ningún antecedente de interés que acude por medios propios describiendo una hematuria secundaria a traumatismo costal derecho mientras jugaba al fútbol. Niega dolor irradiado, no dolor de características mecánicas, no síndrome miccional pero está claramente preocupado por una hematuria macroscópica asintomática. Sin fiebre ni otra clínica concomitante en el resto de anamnesis por aparatos. Antes de su exploración, llama la atención en sus constantes una Tensión Arterial de 185/111 mmHg, frecuencia cardíaca normal (80 lpm), Temperatura 36,4 ºC y SatO2 aire ambiente 98%; respecto al dolor al preguntarle mediante la Escala Visual Analógica lo califica como 3/10.
Buen estado general, normocoloreado, normohidratado, eupneico. Glasgow 15. No impresiona de gravedad
Presenta un abdomen blando, depresible no doloroso y sin defensa abdominal, puñopercusión renal izquierda positiva. No se palpan claras masas ni megalias, no orificios herniarios. No signos de irritación peritoneal. Resto de exploración completamente anodina. Dada clínica, así como antecedente, decidimos canalizar una vía periférica para analgesia endovenosa y analítica sanguínea que pone de manifiesto:
– Bioquímica: glucemia 91 mg/dL, urea 35 mg/dL, Creatinina 1.05mg/dL, iones sin alteraciones.
– Hemograma: Hemoglobina 13 g/dL, Hematocrito 39.2%, leucocitos 11760 con fórmula normal, plaquetas 217000.
– Coagulación sin alteraciones.
Además, solicitamos un sistemático de orina en el que el análisis por citometría no se puede realizar por intensa hematuria. Decidimos revisar sus analíticas previas y llama la atención una Hemoglobina de 12.6g/dL con hematuria microscópica y Sangre Oculta en Heces negativa. Indagando en antecedentes familiares, reconocen problemas renales no filiados en abuelo materno. Durante su observación en Urgencias, tensiones arteriales persistentemente elevadas (en torno a 190/100 mmHg). Comentamos el caso con Urología de guardia quien recomienda TC de abdomen con contraste (imagen 1 y 2):
– Poliquistosis renal bilateral, probablemente autosómica dominante, con quistes hemorrágicos en ambos riñones, más abundantes en el izquierdo.
– Litiasis milimétricas en los tercios superiores y medio del riñón derecho e inferior del riñón izquierdo.
– Vértebra transicional lumbosacra tipo 1A de Castellví.
DISCUSIÓN Y CONCLUSIONES
Los pacientes con Poliquistosis renal autosómica dominante pueden presentar una variedad de condiciones clínicas. La función renal puede permanecer normal durante décadas. Sin embargo, una vez que la tasa de función glomerular comienza a disminuir, la insuficiencia renal suele ser rápida, con una pérdida promedio de 4 a 5 ml/min/año. El sexo masculino, la edad temprana de aparición, el genotipo PKD1 y la proteinuria son indicadores de peor pronóstico8.
Como en este caso, la hipertensión es la presentación clínica más universal y temprana en la mayoría de los pacientes con poliquistosis renal9. La microalbuminuria, la proteinuria y la hematuria también son más prevalentes en estos pacientes. Los episodios de dolor agudo en el flanco a menudo se observan debido a sangrado del quiste, infección, cálculos y (raramente) tumores. La hemorragia del quiste es una complicación frecuente que causa hematuria macroscópica cuando el quiste se comunica con el sistema colector.
No en este caso, pero la prevalencia de quistes hepáticos aumenta con la edad y se debe sospechar enfermedad hepática poliquística cuando hay cuatro o más quistes en el parénquima hepático10. Alrededor del 7 al 36 % de los pacientes también tienen quistes pancreáticos, que son más comunes en la PKD2. mutación11.
Cuando se sospecha PQRAD, la ecografía suele ser suficiente en pacientes asintomáticos con función renal normal. La tomografía computarizada puede ayudar a estimar el volumen renal total ajustado en altura para estratificar el riesgo de progresión de la enfermedad y puede ser beneficiosa para el tratamiento. Según las pautas de práctica clínica de la Asociación Renal, los padres o familiares de personas con poliquistosis renal deben recibir educación sobre el riesgo de heredar PQRAD. La presión arterial debe controlarse cada dos años12.
Finalmente, hablando de tratamiento, tenemos que gestionar todas las complicaciones. Se requiere analgesia cuando un paciente presenta dolor en el flanco o incluso neforlitiasis, los episodios de hemorragia quística suelen ser autolimitados y es fundamental un adecuado manejo de la presión arterial para reducir la mortalidad cardiovascular y la progresión de la insuficiencia renal. Según el estudio HALT-PKD, el rango objetivo de presión arterial es inferior a 120-125/80 mmHg, similar al de otros pacientes con enfermedad renal crónica; sin embargo, en pacientes con función glomerular conservada, un objetivo de presión arterial más bajo (110/75 mmHg) se asocia con una menor incidencia de eventos cardiovasculares y una tasa más lenta de crecimiento de quistes13. Cómo utilizamos en este paciente, los inhibidores de angiotensina son los agentes preferidos si no hay contraindicaciones. La nefrectomía sólo está indicada en pacientes con Poliquistosis renal autosómica dominante con malestar abdominal insoportable, hemorragia renal no controlada, infecciones renales no controladas y carcinoma de células renales.
BIBLIOGRAFÍA
- Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet 2007; 369:1287-301.
- Stengel B, Billon S, Van Dijk PC, et al. Trends in the incidence of renal replacement therapy for end-stage renal disease in Europe, 1990-1999. Nephrol Dial Transplant 2003;18:1824-33.
- Hughes J, Ward CJ, Peral B, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 1995;10:151-60.
- Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int 2009; 76:149-68.
- Pei Y, Obaji J, Dupuis A, Paterson AD, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol 2009;20:205-12.
- Bajwa ZH, Gupta S, Warfield CA, Steinman TI. Pain management in polycystic kidney disease. Kidney Int 2001;60:1631-44.
- Bajwa ZH, Sial KA, Malik AB, Steinman TI. Pain patterns in patients with polycystic kidney disease. Kidney Int 2004;66:1561-9.
- Mahboob M, Rout P, Bokhari SRA. Autosomal Dominant Polycystic Kidney Disease. [Updated 2023 Oct 18]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532934/
- Bell PE, Hossack KF, Gabow PA, Durr JA, Johnson AM, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease. Kidney Int. 1988 Nov;34(5):683-90.
- Gabow PA, Johnson AM, Kaehny WD, Manco-Johnson ML, Duley IT, Everson GT. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990 Jun;11(6):1033-7.
- Kim JA, Blumenfeld JD, Chhabra S, Dutruel SP, Thimmappa ND, Bobb WO, Donahue S, Rennert HE, Tan AY, Giambrone AE, Prince MR. Pancreatic Cysts in Autosomal Dominant Polycystic Kidney Disease: Prevalence and Association with PKD2 Gene Mutations. Radiology. 2016 Sep;280(3):762-70.
- Dudley J, Winyard P, Marlais M, Cuthell O, Harris T, Chong J, Sayer J, Gale DP, Moore L, Turner K, Burrows S, Sandford R. Clinical practice guideline monitoring children and young people with, or at risk of developing autosomal dominant polycystic kidney disease (ADPKD). BMC Nephrol. 2019 Apr 30;20(1):148.
- Schrier RW, Abebe KZ, Perrone RD, Torres VE, Braun WE, Steinman TI, Winklhofer FT, Brosnahan G, Czarnecki PG, Hogan MC, Miskulin DC, Rahbari-Oskoui FF, Grantham JJ, Harris PC, Flessner MF, Bae KT, Moore CG, Chapman AB., HALT-PKD Trial Investigators. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med. 2014 Dec 11;371(24):2255-66.
ANEXOS
IMAGEN 1: Tomografía axial computarizada con contaste. Corte axial.
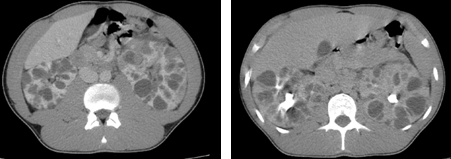
IMAGEN 2: Tomografía axial computarizada con contaste. Corte coronal.



