

Nº de DOI: 10.34896/RSI.2024.34.41.001
AUTORES
- Ana García Esteban. Neumólogo Hospital Clínico, Zaragoza.
- Xunxiao Lin. Neumólogo Hospital Clínico, Zaragoza.
- Lucía Elosúa Prats. Neumólogo Hospital Clínico, Zaragoza.
- Patricia Iñiguez de Heredia Monforte. Neumólogo Hospital Clínico, Zaragoza.
- Guillermo Samuel Loscertales Vacas. Neumólogo Hospital San Pedro, Logroño.
- Marta Martín Lana. Neumólogo Hospital Clínico, Zaragoza.
RESUMEN
La fibrosis pulmonar idiopática (FPI) es una neumonía intersticial fibrosante crónica de causa desconocida, aunque existen factores genéticos, como polimorfismos relacionados con la reparación del epitelio bronquial, y factores ambientales, como el tabaquismo y la exposición laboral, que se asocian con su desarrollo. La disnea de esfuerzo, la tos crónica y la desaturación con el ejercicio son los síntomas más representativos y en la auscultación pulmonar suelen predominar los crepitantes bibasales secos. La FPI se asocia con patrones característicos radiológicos e histológicos de la neumonía intersticial usual (NIU). En ocasiones la sospecha clínica y el patrón radiológico NIU probable o definitivo permite establecer el diagnóstico de FPI. En caso contrario, puede ser necesario confirmarlo mediante una biopsia pulmonar. No existe ninguna medicación que cure la enfermedad, pero dos medicamentos antifibróticos, nintedanib y pirfenidona, han demostrado retrasar la progresión de la enfermedad. Además, en aquellos pacientes que cumplan los criterios necesarios se debería valorar de forma precoz la posibilidad de realizar un trasplante de pulmón. A continuación, se presenta el caso de un paciente diagnosticado de FPI y la evolución natural de su enfermedad a lo largo del tiempo.
PALABRAS CLAVE
Fibrosis pulmonar idiopática, neumonía intersticial usual, nintedanib, pirfenidona.
ABSTRACT
Idiopathic pulmonary fibrosis (IPF) is a chronic fibrosing interstitial pneumonia of unknown cause, although genetic factors, such as polymorphisms related to bronchial epithelial repair, and environmental factors, such as smoking and occupational exposure, are associated with its development. Exertional dyspnoea, chronic cough and desaturation with exercise are the most representative symptoms and dry bibasal crackles usually predominate on lung auscultation. IPF is associated with characteristic radiological and histological patterns of usual interstitial pneumonia (UIP). Sometimes clinical suspicion and a probable or definitive UIP radiological pattern allows the diagnosis of IPF to be established. Otherwise, it may need to be confirmed by lung biopsy. There is no curative medication, but two anti-fibrotic drugs, nintedanib and pirfenidone, have been shown to slow disease progression. In addition, lung transplantation should be considered early in patients who meet the necessary criteria. Next, we present the case of a patient diagnosed with IPF and the natural progression of his disease over time.
KEY WORDS
Idiopathic pulmonary fibrosis, usual interstitial pneumonia, nintedanib, pirfenidone.
INTRODUCCIÓN
Las enfermedades pulmonares intersticiales difusas (EPID) constituyen un grupo heterogéneo de enfermedades, caracterizadas por la afectación del intersticio pulmonar y, en ocasiones, también del epitelio alveolar y vascular. En las EPID se encuentran involucrados procesos de reparación, con distintos grados de inflamación y de fibrosis. La fibrosis es un proceso fisiológico de reparación del tejido dañado que consiste en generar un tejido de mayor resistencia. Sin embargo, daños repetidos pueden conllevar un mayor grado de fibrosis que desencadene un mal funcionamiento del tejido1.
La fibrosis pulmonar idiopática (FPI) es la EPID más frecuente según la mayoría de estudios epidemiológicos, con una prevalencia estimada de 12 casos por cada 100.000 mujeres y 20 casos por cada 100.000 hombres2.
Se trata de una neumonía intersticial fibrosante crónica de aparición espontánea y de causa desconocida (idiopática) caracterizada por una sustitución difusa del parénquima pulmonar por un depósito de matriz extracelular que conduce a una fibrosis irreversible3. Además, aunque la mayoría de los casos de FPI son esporádicos, existe evidencia que respalda la presencia de un componente genético predisponente. Se ha documentado la presencia de diversos polimorfismos genéticos relacionados con la reparación del epitelio bronquial: proteínas A2 y C del surfactante, mucinas (polimorfismo de la MUC5B) y el complejo telomerasa (genes TERC y TERT) vinculados al acortamiento de los telómeros y a la dificultad en la reparación tisular y del daño del ADN4.
Además, también se han descrito ciertos factores de riesgo. El tabaquismo, la exposición laboral a piedra, metal, madera, polvos orgánicos y contaminantes del aire está estrechamente relacionado con al FPI5.
La edad media de diagnóstico se sitúa en torno a los 65 años, siendo muy raro en pacientes menores de 50 años4. Suele ser más frecuente en varones y, además, existen factores de riesgo para su aparición, entre los que se encuentra el tabaquismo, los contaminantes ambientales, la exposición ocupacional (polvos de madera, metálicos, agricultura, ganadería…) o el reflujo gastroesofágico6.
PRESENTACIÓN DEL CASO CLÍNICO
Se trata de un paciente varón de 64 años, sin alergias medicamentosas conocidas, exfumador de 1 paquete y medio al día durante 40 años (IPA de 60 paquetes/año). Jubilado, previamente trabajaba en soldadura.
Derivado desde Atención Primaria por disnea grado 1 según la escala mMRC y escasa tos de predominio matutino con expectoración mucosa. A la exploración, se objetivan acropaquias en ambas manos y a la auscultación pulmonar presenta hipofonesis generalizada sin ruidos patológicos sobreañadidos.
Se solicita estudio funcional respiratorio, con espirometría normal y un descenso moderado de la difusión de monóxido de carbono. Además, se solicita un TACAR de tórax (Imagen 1), en el que se visualiza enfisema pulmonar paraseptal y centrolobulillar avanzado, así como bronquiectasias periféricas bibasales con afectación reticular, con moderados signos de fibrosis pulmonar, todo ello en el contexto de un patrón de probable NIU.
Se completa el estudio solicitando un test de la marcha de 6 minutos, en el que el paciente recorre una distancia de 240 metros, que equivale a un 49% de su valor de referencia.
Basándonos en la sospecha clínica y la información aportada por las pruebas complementarias solicitadas, se establece el diagnóstico de fibrosis pulmonar idiopática sobre enfisema pulmonar y se mantiene un seguimiento periódico anual de la evolución clínica y funcional del paciente.
Tras 5 años de seguimiento, se comprueba la progresividad y el empeoramiento funcional de la enfermedad (Imagen 2), por lo que se plantea añadir un fármaco antifibrótico y se decide iniciar tratamiento con nintedanib (1 comprimido de 150 mg cada 12 horas) con control analítico trimestral de transaminasas. Como efecto adverso el paciente presenta molestias digestivas y diarreas, que se resuelven tras el inicio de dieta astringente y el tratamiento con harina de algarrobo y ultralevura.
DISCUSIÓN-CONCLUSIONES
En la fibrosis pulmonar idiopática, los síntomas respiratorios suelen ser inicialmente leves e inespecíficos. No obstante, la disnea de esfuerzo, la tos crónica no productiva y la desaturación con el ejercicio constituyen los síntomas más representativos. En la auscultación pulmonar suelen predominar los crepitantes teleinspiratorios bibasales secos y durante la exploración del paciente podemos detectar la presencia de acropaquias, aunque este último hallazgo suele aparecer en fases avanzadas de la enfermedad4.
A nivel de la mecánica pulmonar, estos pacientes presentan una disminución de la distensibilidad, de tal manera que son necesarias presiones muy altas para producir un cambio en el volumen pulmonar. Esto se traduce, en el estudio funcional respiratorio, en forma de un patrón restrictivo con disminución de la capacidad vital forzada (CVF) y un volumen espiratorio forzado en el primer segundo (FEV1) reducido, con una relación FEV1/FVC normal o aumentada, así como una disminución precoz de la capacidad de difusión del monóxido de carbono (DLCO)6.
La fibrosis pulmonar idiopática se asocia con patrones característicos radiológicos e histológicos de la neumonía intersticial usual (NIU), aunque no son específicos de esta enfermedad4. En la tomografía computarizada de alta resolución (TCAR) este patrón se caracteriza por la presencia de bronquiectasias/bronquiectasias por tracción y opacidades periféricas de predominio basal y periférico asociadas a panalización de localización subpleural. La formación de panal de abeja confirma de forma definitiva el diagnóstico de NIU, mientras que las bronquiectasias por tracción por sí solas indican una NIU probable. También hay que tener en cuenta que durante una exacerbación aguda de FPI pueden aparecer opacidades bilaterales en vidrio deslustrado7.
En cuanto al diagnóstico, ninguna prueba de laboratorio es específica para el diagnóstico de FPI, por lo que su función es identificar y excluir otros procesos en el diagnóstico diferencial. Habitualmente se debe solicitar una analítica con perfil de autoinmunidad para descartar una enfermedad reumática subclínica (artritis reumatoide, miositis, esclerodermia…). Otra prueba que puede resultar útil para descartar diagnósticos alternativos es el lavado broncoalveolar (BAL), sobre todo se recomienda cuando las imágenes radiológicas son indeterminadas u orientan a otras patologías4.
Es importante tener en cuenta que las directrices de 2022 de la Sociedad Torácica Americana (ATS), la Sociedad Respiratoria Europea (ERS), la Sociedad Respiratoria Japonesa (JRS) y la Sociedad Torácica Latinoamericana (ALAT) sugieren que una biopsia de pulmón no es necesaria en pacientes con un patrón NIU de TCAR considerado definitivo o probable4. Sin embargo, cuando la evaluación clínica, las pruebas complementarias solicitadas y la discusión multidisciplinar del caso no nos permiten establecer un diagnóstico seguro de FPI, debemos considerar confirmar el diagnóstico mediante una biopsia pulmonar, siempre teniendo en cuenta las comorbilidades de nuestro paciente y la relación riesgo-beneficio4.
Además, debemos tener en cuenta que la muestra obtenida debe ser lo suficientemente grande, ya sea mediante biopsia pulmonar quirúrgica (toracotomía o videotoracoscopia) o, como alternativa en centros con experiencia, mediante criobiopsia transbronquial4. El estudio histopatológico muestra una afectación heterogénea con inflamación intersticial, áreas en panal de abeja y focos fibroblásticos adyacentes a células epiteliales alveolares hiperplásicas alternando con áreas de pulmón normal6.
Respecto al tratamiento, no existe ningún medicamento que cure la enfermedad, pero dos medicamentos antifibróticos, nintedanib y pirfenidona, parecen retrasar la progresión de la enfermedad, disminuir la tasa de exacerbaciones y el riesgo de mortalidad por todas las causas8. Ambos medicamentos exigen un control estrecho de la función hepática, ya que uno de sus efectos secundarios más frecuente es la hepatotoxicidad, con la consiguiente elevación de los niveles séricos de transaminasas6.
No obstante, dado el carácter progresivo de esta enfermedad, en estos pacientes se debería valorar de forma precoz la posibilidad de realizar un trasplante de pulmón, incluso antes de comprobar la respuesta al tratamiento médico pautado, siempre y cuando no haya contraindicaciones y el paciente cumpla los criterios necesarios para incluirlo en la lista de trasplantes9.
Aunque a día de hoy no existe ningún tratamiento curativo para la FPI, se están llevando a cabo ensayos clínicos prometedores con nuevos fármacos que podrían suponer un cambio significativo en la historia natural de esta enfermedad.
BIBLIOGRAFÍA
- Liebow A, Carrington C.B. The Interstitial Pneumonias. In: Simon M., Potchen E.J., Lemay E., editors. Frontiers in pulmonary radiology. New York: Grune and Stratton; 1969; 102–141.
- Podolanczuk AJ, Thomson CC, Remy-Jardin M, et al. Idiopathic pulmonary fibrosis: state of the art for 2023. Eur Respir J. 2023; 61(4).
- Wijsenbeek M, Suzuki A and Maher TM. Interstitial lung diseases. Lancet. 2022; 400(10354): 769-786.
- Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022; 205(9).
- Baumgartner KB, Samet JM, Coultas DB, et al. Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case-control study. Collaborating Centers. Am J Epidemiol 2000; 152:307.
- Glass DS, Grossfeld D, Renna HA, Agarwala P, Spiegler P, DeLeon J, Reiss AB. Idiopathic pulmonary fibrosis: Current and future treatment. Clin Respir J. 2022;16(2):84-96.
- Hodnett PA, Naidich DP. Fibrosing interstitial lung disease. A practical high-resolution computed tomography-based approach to diagnosis and management and a review of the literature. Am J Respir Crit Care Med 2013; 188:141.
- Dempsey TM, Thao V, Helfinstine DA Jr, et al. Real-world cohort evaluation of the impact of the antifibrotics in patients with idiopathic pulmonary fibrosis. Eur Respir J 2023; 62.
- Weill D, Benden C, Corris PA, et al. A consensus document for the selection of lung transplant candidates: 2014-an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2015; 34:1.
ANEXOS
Imagen 1. TACAR de tórax.
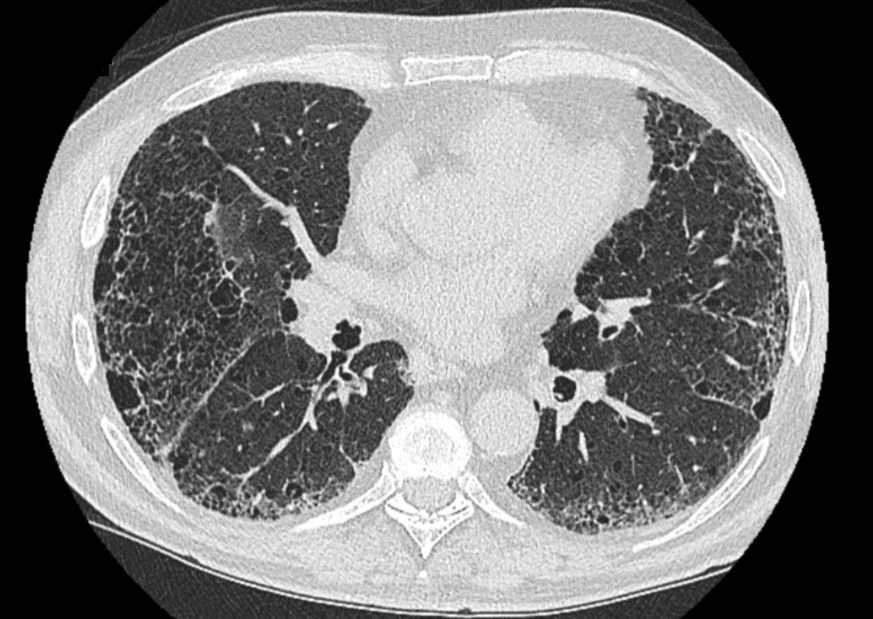
Imagen 2. Progresión del estudio funcional respiratorio.
| Año | FVC | FEV1 | FEV1/FVC | TLC | DLCO | DLCO/VA | pO2 | Sat O2% |
|---|---|---|---|---|---|---|---|---|
| 2018 | 4.21 (92%) | 3.20 (94%) | 76% | 4.95 (55%) | 68% | 66 | 93 | |
| 2019 | 3.94 (86%) | 2.86 (85%) | 73% | 5.39 (83%) | 5.67 (64%) | 74% | 74 | 96 |
| 2020 | 3.85 (85%) | 2.60 (79%) | 68% | 5.98 (92%) | 4.84 (56%) | 75% | 79 | 96 |
| 2021 | 3.23 (81%) | 2.19 (71%) | 68% | 4.88 (75%) | 3.96 (46%) | 69% | 76 | 96 |
| 2022 | 3.34 (84%) | 2.35 (76%) | 70% | 4.78 (73%) | 3.50 (41%) | 49% | 74 | 95 |
| 2023 | 3.67 (93%) | 2.50 (82%) | 68% | 5.05 (77%) | 3.23 (38%) | 54% | 81 | 96 |
Imagen 3. Progresión de prueba de la marcha de seis minutos.
| Fecha | Metros | Sat. O2 inicial | Sat. O2 final | FC Inicial | FC Final | Borg previo | Borg final | Fatiga EEII previo | Fatiga EEII final |
|---|---|---|---|---|---|---|---|---|---|
| 2021 (sin oxígeno) | 240 (49%) | 91 | 82 | 106 | 99 | 0 | 4 | 0 | 0 |
| 2021 (con oxígeno) | 350 (71%) | 93 | 86 | 108 | 120 | 0 | 2 | 0 | 0 |
| 2022 (sin oxígeno) | 420 (86%) | 94 | 86 | 91 | 110 | 2 | 3 | 0 | 0 |
| 2022 (con oxígeno) | 430 (88%) | 94 | 88 | 100 | 107 | 2 | 3 | 0 | 0 |
| 2023 (sin oxígeno) | 386 (79%) | 93 | 86 | 100 | 107 | 0 | 3 | 0 | 0 |


