

Nº de DOI: 10.34896/RSI.2024.15.87.001
AUTORES
- Lucía Elosúa Prats. Neumóloga Hospital Clínico, Zaragoza, España.
- Carlos Murillo Arribas. Neumólogo Hospital Clínico, Zaragoza, España.
- Guillermo Samuel Loscertales Vacas. Neumólogo Hospital San Pedro, Logroño, España.
- Marta Martín Lanas. Neumóloga Hospital Clínico, Zaragoza, España.
- Ana García Esteban. Neumóloga Hospital Clínico, Zaragoza, España.
- Xunxiao Lin. Neumóloga Hospital Clínico, Zaragoza, España.
RESUMEN
La linfangioleiomiomatosis (LAM) es una enfermedad poco común que afecta principalmente a mujeres jóvenes y puede presentarse con neumotórax espontáneo, disnea inexplicable, colecciones de líquido quiloso pleural o peritoneal, o asociada al complejo de esclerosis tuberosa (CET) o angiomiolipoma renal (AML). La evaluación diagnóstica incluye pruebas de función pulmonar y tomografía computarizada de alta resolución para detectar quistes pulmonares característicos. Además, se pueden realizar pruebas serológicas, como la medición del factor de crecimiento endotelial vascular sérico D (VEGF-D), y exámenes abdominopélvicos para evaluar la presencia de angiomiolipomas renales (AML). La biopsia pulmonar se reserva para casos atípicos o cuando no se cumplen los criterios de diagnóstico clínico. El diagnóstico diferencial incluye otras enfermedades pulmonares quísticas, neumotórax y quilotórax. Es crucial evaluar la extensión de la enfermedad para guiar el tratamiento y el seguimiento adecuados, ya que la LAM es progresiva y multisistémica.
A continuación, se presenta el caso de una paciente mujer joven con clínica de disnea progresiva junto con derrame pleural a estudio. Se llevó a cabo una serie de pruebas complementarias llegando al diagnóstico clínico definitivo de LAM esporádica.
PALABRAS CLAVE
Linfangioleiomiomatosis, quilotórax, tomografía computarizada, quistes pulmonares, biopsia pulmonar.
ABSTRACT
Lymphangioleiomyomatosis (LAM) is a rare disease that mainly affects young women and may present with spontaneous pneumothorax, unexplained dyspnea, pleural or peritoneal chylous fluid collections, or in association with tuberous sclerosis complex (TSC) or renal angiomyolipoma (AML). Diagnostic evaluation includes pulmonary function tests and high-resolution computed tomography to detect characteristic lung cysts. In addition, serological tests, such as measurement of serum vascular endothelial growth factor D (VEGF-D), and abdominopelvic examinations to assess for the presence of renal angiomyolipomas (AML) may be performed. Lung biopsy is reserved for atypical cases or when clinical diagnostic criteria are not met. The differential diagnosis includes other cystic lung diseases, pneumothorax and chylothorax. It is crucial to assess the extent of the disease to guide appropriate treatment and follow-up, as LAM is progressive and multisystemic.
Below is the case of a young female patient with progressive dyspnea and pleural effusion under study. A series of complementary tests were carried out leading to a definitive clinical diagnosis of sporadic LAM.
KEY WORDS
Lymphangioleiomyomatosis, chylothorax, computed tomography, pulmonary cysts, lung biopsy.
INTRODUCCIÓN
La linfangioleiomiomatosis (LAM) es una enfermedad multisistémica poco frecuente que afecta principalmente a mujeres jóvenes y los síntomas varían dependiendo de los órganos afectados. Además de los pulmones, la enfermedad puede manifestarse en los riñones y la vasculatura linfática1. A diferencia de la esclerosis tuberosa, la LAM esporádica no presenta síntomas neurocutáneos típicos. Mayormente afecta a mujeres premenopáusicas, con inicio promedio entre los treinta y cuarenta años, siendo más común en mujeres blancas de estatus socioeconómico elevado2.
Los síntomas pulmonares abarcan fatiga, dificultad para respirar, neumotórax espontáneo, derrame pleural, dolor torácico, tos y hemoptisis, siendo la dificultad respiratoria durante el ejercicio el síntoma más prevalente3. Algunas pacientes experimentan una exacerbación cíclica de los síntomas relacionada con su ciclo menstrual4.
Los angiomiolipomas renales (AML) son frecuentes en la LAM y pueden ser asintomáticos o provocar hemorragias y dolor abdominal en casos graves. Se estima que afectan alrededor del 30% de los individuos con LAM esporádica, en contraste con más del 80% de aquellos con CET-LAM5.
Las manifestaciones linfáticas, como el quilotórax y el quiloperitoneo, son comunes, al igual que la LAM retroperitoneal y la linfadenopatía, siendo más prevalentes en la LAM esporádica que en la asociada a la esclerosis tuberosa. Según investigaciones, se observa que los derrames quilosos tienden a presentarse mayormente de forma unilateral (aproximadamente un 76%) y preferentemente en el lado derecho; no obstante, es posible que también se desarrollen derrames bilaterales6.
Cuando se sospecha LAM, es crucial realizar una evaluación diagnóstica completa que incluya: pruebas de función pulmonar (espirometría y capacidad de difusión de monóxido de carbono (DLCO)), aunque unos resultados normales no excluyen el diagnóstico; Tomografía computarizada de alta resolución (TACAR) sin contraste para detectar los quistes pulmonares característicos de la enfermedad; examen completo para detectar síntomas y signos del complejo de esclerosis tuberosa (CET), ya que la LAM puede estar asociada con este trastorno; imágenes abdominopélvicas para buscar la presencia de AML, derrames quilosos, linfadenopatía y/o linfangioleiomiomas; medición del factor de crecimiento endotelial vascular sérico D (VEGF-D), donde un nivel ≥800 pg/mL ayuda a distinguir la LAM de otras enfermedades pulmonares quísticas7,8,9.
PRESENTACIÓN DEL CASO CLÍNICO
Se trata de una mujer de 36 años, natural de Rumanía, residente en España desde hace 5 años. Fumadora activa de un paquete al día desde hace 10 años (IPA 10). Trabaja como teleoperadora. Vive en domicilio con su marido, no animales domésticos.
Síndrome de Raynaud como único antecedente médico de interés. No tratamiento crónico habitual.
Historial actual: valorada el día 13 de febrero en su Centro de Salud por clínica de aproximadamente 2 semanas de inicio con cuadro de tos no productiva, sin fiebre, valorado como cuadro catarral iniciando tratamiento sintomático (paracetamol, mucolítico). Acude de nuevo a Urgencias del Hospital Clínico el día 17 de febrero por empeoramiento del cuadro clínico con disnea de esfuerzo en aumento, empeoramiento de la tos, ahora con expectoración escasa amarillenta y dolor torácico referido a plano posterior de ambos campos pulmonares sin traumatismo previo. Termometrada sin fiebre. No sensación distérmica. SatO2 95% basal. Entre las pruebas complementarias realizadas, destaca en analítica sanguínea ligera leucocitosis con desviación izquierda y en radiografía de tórax derrame pleural derecho en moderada cuantía, por lo que es ingresada en Neumología para completar estudio.
Exploración física: A su llegada a planta hospitalaria, consciente y orientada, afebril, hemodinámicamente estable, eupneica en reposo con buena saturación de oxígeno, sin focalidad neurológica. No se palpan adenopatías. La auscultación cardíaca revela ritmos cardíacos rítmicos y regulares. La auscultación pulmonar muestra hipoventilación hasta campos medios de hemitórax derecho con normoventilación en hemitórax izquierdo. La exploración abdominal es anodina, y no se detectan edemas en las extremidades inferiores.
Resumen de pruebas diagnósticas:
- Se realiza ecografía torácica en hemitórax derecho con la paciente en sedestación, objetivando líquido pleural anecoico de elevada cuantía sin ecos en su interior y que ocasiona colapso del parénquima pulmonar subyacente. Se localiza punto de punción y se realiza toracocentesis diagnóstica con extracción de 25 cc de líquido pleural con aspecto macroscópico lechoso sugestivo de posible quilotórax, por lo que se añade a la bioquímica triglicéridos, colesterol total y quilomicrones.
- Muestra Líquido pleural: aspecto lechoso. Triglicéridos 2688 mg/dL (>110 mg/dL) y colesterol 80.6 mg/dL (<200 mg/dL). Citología de líquido pleural: leve componente inflamatorio.
- TC de tórax de alta resolución sin contraste intravenoso: Marcado derrame pleural derecho con atelectasia del parénquima subyacente que provoca desplazamiento contralateral mediastínico. No se visualizan adenomegalias hiliomediastínicas. No se visualiza derrame pericárdico. No se aprecian nódulos o masas pulmonares. Múltiples quistes de pequeño tamaño, bilaterales y difusos, bien definidos, más numerosos en lóbulos superiores y lóbulo medio. Valorar patología intersticial tipo linfangioleiomiomatosis, histiocitosis de células de Langerhans, neumonía intersticial linfocítica…. Marcada escoliosis dorsolumbar. Espondilosis dorsal.
Diagnóstico: ante mujer joven con disnea progresiva, derrame pleural compatible con quilotórax, TAC de tórax con quistes bilaterales y sin historia ni antecedentes de esclerosis tuberosa, se hizo el diagnóstico clínico de linfangioleiomiomatosis esporádica.
Evolución: la paciente tras diagnóstico de quilotórax no traumático y TC de tórax compatible con LAM, fue valorada por parte de Cirugía torácica quien decidió manejo conservador de derrame pleural quiloso sin colocación de tubo de drenaje torácico dada estabilidad respiratoria. Además, fue valorada por parte de Endocrino y Nutrición para inicio de dieta específica. Durante su ingreso, mejoría de la sintomatología con menos sensación disneica y buena saturación de oxígeno sin requerimientos de oxígenos, por lo que fue dada de alta con revisión en consultas de Neumología a los 3 meses con estudio de función respiratoria, analítica sanguínea y radiografía de tórax, y se inició pauta con Sirolimus 1 mg al día.
Tratamiento: dieta vegetariana y ácidos grasos de cadena media. Inhibidor mTOR (Sirolimus).
DISCUSIÓN-CONCLUSIONES
La linfangioleiomiomatosis (LAM) es una enfermedad multisistémica poco común que afecta principalmente a mujeres jóvenes. El término LAM esporádica se aplica a pacientes sin complejo de esclerosis tuberosa (CET)1.
La LAM debe sospecharse en mujeres jóvenes con neumotórax espontáneo, disnea inexplicable, y/o colecciones de líquido quiloso3. Los quistes pulmonares característicos de la LAM se encuentran comúnmente en la TC, incluso en pacientes asintomáticos7.
Las conclusiones clave de este caso clínico serían las siguientes:
Sospecha Clínica de LAM:
Mujer joven con disnea progresiva y derrame pleural. Sin antecedentes médicos de interés. Niega traumatismo torácico previo.
La toracocentesis diagnóstica mostró un líquido pleural con aspecto macroscópico lechoso sugestivo de quilotórax.
Hallazgos Radiológicos y de Laboratorio:
El análisis bioquímico de líquido pleural fue diagnóstico definitivo de quilotórax no traumático (triglicéridos 2688 mg/dL (>110 mg/dL) y colesterol 80.6 mg/dL (<200 mg/dL))10.
La tomografía computarizada de alta resolución (TACAR) de tórax reveló quistes de pequeño tamaño, de manera difusa, bilateral y bien definidos. Hallazgos radiológicos sugestivos de linfangioleiomiomatosis7.
Diagnóstico clínico de LAM:
El diagnóstico de LAM puede establecerse mediante TACAR del tórax cuando se identifican quistes característicos en pacientes con complejo de esclerosis tuberosa (CET), angiomiolipomas renales (AML), niveles elevados de factor de crecimiento endotelial vascular D (VEGF-D; ≥800 pg/mL), acumulaciones de quilo y/o la apariencia típica de linfangioleiomiomas en la TC11.
En nuestro caso, se realizó un diagnóstico clínico de LAM esporádica.
Diagnóstico patológico definitivo mediante biopsia pulmonar:
La necesidad de biopsia depende de los hallazgos clínicos y radiológicos. En algunos casos, se puede prescindir de la biopsia si los criterios clínicos y de imagen son concluyentes para el diagnóstico.
Cuando se evalúa la posibilidad de diagnóstico de LAM, el objetivo principal es alcanzar el diagnóstico más concluyente empleando la estrategia menos invasiva disponible como se ha mencionado en el punto anterior. Estas estrategias reducen la necesidad de realizar biopsias y son adecuadas para respaldar el uso de inhibidores mTOR en el tratamiento12.
Evaluación de la extensión:
La LAM es una enfermedad progresiva y multisistémica que requiere una evaluación inicial exhaustiva de su extensión, seguida de un seguimiento longitudinal para monitorear su progresión y complicaciones. Se recomienda realizar pruebas diagnósticas como imágenes abdominopélvicas para evaluar completamente los angiomiolipomas renales y extrarrenales, así como otros hallazgos como linfadenopatías y linfangioleiomiomas. Además, se sugiere evaluar la capacidad de ejercicio y realizar pruebas dirigidas a los síntomas específicos, como la hipertensión pulmonar y la osteoporosis. Los niveles de VEGF-D también pueden ser útiles como biomarcadores pronósticos, aunque su papel en las decisiones de tratamiento aún se está investigando. Se aconseja realizar pruebas de detección para el cáncer y la osteoporosis según sea necesario. Además, se recomienda realizar pruebas frecuentes de la función pulmonar durante el seguimiento para evaluar la progresión de la enfermedad pulmonar con precisión13.
En conclusión, la LAM es una enfermedad poco común que afecta principalmente a mujeres jóvenes y se caracteriza por su naturaleza multisistémica. El diagnóstico de LAM esporádica se establece en pacientes sin complejo de esclerosis tuberosa (CET) y se sospecha en aquellos con neumotórax espontáneo, disnea inexplicable y/o colecciones de líquido quiloso. Los hallazgos radiológicos y de laboratorio, como la presencia de quilotórax no traumático y quistes pulmonares característicos en la TACAR, respaldan el diagnóstico clínico de LAM y son suficientes para iniciar tratamiento. La necesidad de biopsia para un diagnóstico patológico definitivo depende de los hallazgos clínicos y de imagen, y se busca alcanzar el diagnóstico más concluyente con la estrategia menos invasiva disponible. La evaluación de la extensión de la enfermedad incluye pruebas adicionales y seguimiento longitudinal para monitorear la progresión y complicaciones.
BIBLIOGRAFÍA
- Matsui K, Tatsuguchi A, Valencia J, et al. Extrapulmonary lymphangioleiomyomatosis (LAM): clinicopathologic features in 22 cases. Hum Pathol 2000; 31:1242.
- Ryu JH, Moss J, Beck GJ, et al. The NHLBI lymphangioleiomyomatosis registry: characteristics of 230 patients at enrollment. Am J Respir Crit Care Med 2006; 173:105.
- Corrin B, Liebow AA, Friedman PJ. Pulmonary lymphangiomyomatosis. A review. Am J Pathol 1975; 79:348.
- Shaw BM, Kopras E, Gupta N. Menstrual Cycle-related Respiratory Symptom Variability in Patients with Lymphangioleiomyomatosis. Ann Am Thorac Soc 2022; 19:1619.
- Bernstein SM, Newell JD Jr, Adamczyk D, et al. How common are renal angiomyolipomas in patients with pulmonary lymphangiomyomatosis? Am J Respir Crit Care Med 1995; 152:2138.
- Ryu JH, Doerr CH, Fisher SD, et al. Chylothorax in lymphangioleiomyomatosis. Chest 2003; 123:623.
- Gupta N, Finlay GA, Kotloff RM, et al. Lymphangioleiomyomatosis Diagnosis and Management: High-Resolution Chest Computed Tomography, Transbronchial Lung Biopsy, and Pleural Disease Management. An Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guideline. Am J Respir Crit Care Med 2017; 196:1337.
- Hayashida M, Yasuo M, Hanaoka M, et al. Reductions in pulmonary function detected in patients with lymphangioleiomyomatosis: An analysis of the Japanese National Research Project on Intractable Diseases database. Respir Investig 2016; 54:193.
- Cottin V, Lacronique J, Denis L, et al. Diagnostic value of serum VEGF-D in consecutive patients with lymphangioleiomyomatosis. Eur Respir J 2011; 38:627.
- Ryu JH, Doerr CH, Fisher SD, et al. Chylothorax in lymphangioleiomyomatosis. Chest 2003; 123:623.
- Chang WY, Cane JL, Blakey JD, et al. Clinical utility of diagnostic guidelines and putative biomarkers in lymphangioleiomyomatosis. Respir Res 2012; 13:34.
- Gupta N, Finlay GA, Kotloff RM, et al. Lymphangioleiomyomatosis Diagnosis and Management: High-Resolution Chest Computed Tomography, Transbronchial Lung Biopsy, and Pleural Disease Management. An Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guideline. Am J Respir Crit Care Med 2017; 196:1337.
- Baldi BG, Araujo MS, Freitas CS, et al. Evaluation of the extent of pulmonary cysts and their association with functional variables and serum markers in lymphangioleiomyomatosis (LAM). Lung 2014; 192:967.
ANEXOS
IMAGEN 1. Radiografía de tórax: Derrame pleural derecho en moderada cuantía, sin poder descartar afectación en el parénquima subyacente.

IMAGEN 2. Ecografía torácica: líquido pleural anecoico de elevada cuantía sin ecos en su interior y que ocasiona colapso del parénquima pulmonar subyacente.
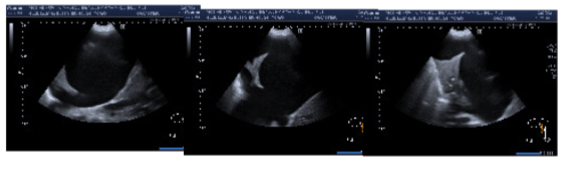
IMAGEN 3. Líquido pleural con aspecto macroscópico lechoso sugestivo de posible quilotórax.



