Nº de DOI: 10.34896/RSI.2024.66.70.001
AUTORES
- Marta Martín Lana. Hospital Clínico Universitario Lozano Blesa, Zaragoza, España.
- Guillermo Samuel Loscertales Vacas. Hospital San Pedro, Logroño, España.
- Xunxiao Lin. Hospital Clínico Universitario Lozano Blesa, Zaragoza, España.
- Ana García Esteban. Hospital Clínico Universitario Lozano Blesa, Zaragoza, España.
- Lucía Elosua Prats. Hospital Clínico Universitario Lozano Blesa, Zaragoza, España.
- Carlos Murillo Arribas. Hospital Clínico Universitario Lozano Blesa, Zaragoza, España.
RESUMEN
La evaluación de la enfermedad pulmonar intersticial (EPI) en pacientes con enfermedades del tejido conectivo (ETC) representa un desafío debido a la heterogeneidad, la presencia de diversos patrones radiológicos y la variabilidad en los grados de gravedad de presentación. Dado que la EPI puede manifestarse en cualquier momento, resulta crucial realizar una evaluación exhaustiva y multidisciplinaria para detectar posibles ETC subyacentes.
La esclerosis sistémica progresiva (ESP) es una enfermedad autoinmune multisistémica crónica caracterizada por el depósito excesivo de colágeno con alteraciones dermatológicas progresivas y afectación de otros órganos diana manifestándose con fibrosis cutánea y visceral. La EPI se asocia a una morbilidad y mortalidad significativas y es la complicación más frecuente de la ESP, de manera que su identificación precoz es crucial para instaurar un tratamiento adecuado.
Se deben realizar estudios de la función respiratoria completos y una tomografía computarizada de alta resolución (TCAR) ante la mínima sospecha de EPI. El tratamiento mediante inmunosupresores para el control de la enfermedad de base, se recomienda en aquellos casos en los que exista un deterioro clínico y funcional, ya que pueden retrasar el progreso de la EPI.
PALABRAS CLAVE
Enfermedades pulmonares intersticiales, enfermedades del tejido conectivo, esclerosis sistémica progresiva.
ABSTRACT
The assessment of interstitial lung disease (ILD) in patients with connective tissue diseases (CTD) represents a challenge due to heterogeneity, diverse radiological patterns, and variability in severity of presentation. As ILD can manifest at any time, a comprehensive and multidisciplinary evaluation for underlying CTD is crucial.
Progressive systemic sclerosis (PSS) is a chronic multisystem autoimmune disease characterised by excessive collagen deposition with progressive dermatological changes and involvement of other target organs manifesting with cutaneous and visceral fibrosis. ILD is associated with significant morbidity and mortality and is the most common complication of PSS, so early identification is crucial for appropriate treatment.
Complete respiratory function studies and high-resolution computed tomography (HRCT) should be performed at the slightest suspicion of ILD. Immunosuppressive treatment to control the underlying disease is recommended in those cases where there is clinical and functional deterioration, as it may delay the progression of ILD.
KEY WORDS
Interstitial lung diseases, connective tissue diseases, progressive systemic sclerosis.
INTRODUCCIÓN
Las enfermedades del colágeno o enfermedades del tejido conectivo (ETC) engloban un amplio grupo de enfermedades caracterizadas por inflamación crónica y/o fibrosis mediada por un mecanismo patogénico autoinmune, que conduce a daños tisulares, depósito de colágeno y posible pérdida de la función del órgano afectado.
La enfermedad pulmonar intersticial (EPI) es una manifestación frecuente de las ETC y a menudo se asocia a una morbilidad y mortalidad significativas, como ocurre en la esclerodermia1. Puede ser causa de la expresión de la propia enfermedad o ser la causa de una complicación de la ETC. La EPI se puede diagnosticar simultáneamente o posteriormente a una ETC conocida, también puede presentarse como la primera manifestación de una ETC previamente no reconocida2.
La tomografía computarizada de alta resolución (TCAR) se considera la principal herramienta diagnóstica para evaluar a los pacientes con ETC, ya que tiene una sensibilidad superior a los estudios convencionales del tórax al revelar de manera precisa los hallazgos macroscópicos y la distribución de la enfermedad pulmonar. En determinadas situaciones clínicas, los hallazgos del TCAR pueden sugerir un diagnóstico específico3,4.
La esclerodermia sistémica progresiva (ESP) es una enfermedad autoinmune multisistémica crónica caracterizada por un depósito de colágeno excesivo que afecta a la piel, los vasos sanguíneos y varios órganos. La EPI es la complicación más común asociada a la ESP, presente en más del 75% de los casos. A menudo, la EPI es subclínica en los estadios precoces de la enfermedad, pero a menudo se encuentran alteraciones en las pruebas funcionales respiratorias (PFR), TCAR y en el análisis del lavado broncoalveolar (LBA). Identificar de manera temprana a los pacientes con EPI es crucial para poder instaurar un tratamiento adecuado5.
LA ESP generalmente suele tener un curso clínico menos agresivo y un mejor pronóstico que la fibrosis pulmonar idiopática. Se asocia a la presencia de ganglios mediastínicos aumentados, vasculitis pulmonares e hipertensión pulmonar. La dilatación esofágica se ha puesto de manifiesto en estudios con tomografía computarizada en el 80% de los pacientes con EPI6.
Los hallazgos en el TCAR de fibrosis intersticial de la ESP incluyen áreas de atenuación en “vidrio deslustrado” asociadas con una infiltración predominantemente celular en la biopsia (NINE) en comparación con el patrón reticular con panalización subpleural con bronquiectasias de tracción, derrame o engrosamiento pleural, todo ello, de distribución preferentemente periférica y basal asociado con una biopsia predominantemente fibrótica (NIU). La forma más frecuente es la NINE, con predominio de la variante fibrótica7, en la que predomina el patrón reticular periférico de predominio basal y opacidades en vidrio deslustrado8.
La base del tratamiento en todas las formas de EPI secundarias a ETC son medicamentos inmunosupresores, como micofenolato de mofetilo, ciclofosfamida o azatioprina. El nintedanib es un medicamento antifibrótico que puede retrasar el progreso de la EPI asociada a esclerodermia9. Estos medicamentos quedan reservados únicamente para aquellos pacientes con enfermedad progresiva y/o clínicamente significativa.
PRESENTACIÓN DEL CASO CLÍNICO
Mujer de 83 años, sin alergias medicamentosas conocidas. Entre sus antecedentes personales destaca hipertensión arterial y diabetes mellitus tipo 2. Independiente para las actividades básicas de la vida diaria. Vive en domicilio con su marido y un hijo. Ha trabajado de costurera y ama de casa. Tiene como mascota un perro. Nunca fumadora, sin exposición a tóxicos ni otros neumoalérgenos.
Acude a urgencias de su hospital por disnea de una semana de evolución hasta hacerse de mínimos esfuerzos. Al interrogatorio confirma disnea basal grado 3 mMRC de aproximadamente tres años de evolución. Refiere tos con expectoración mucopurulenta y debilidad generalizada. No refiere fiebre en domicilio, pero a su llegada a urgencias se objetiva una temperatura de 39ºC. No dolor torácico. No signos de insuficiencia cardiaca. No clínica digestiva ni miccional. Pérdida ponderal de años de evolución. Niega ambiente epidemiológico familiar.
En la exploración física estabilidad hemodinámica con saturación de oxígeno basal de 87%, precisando de oxigenoterapia a flujos bajos. A la auscultación pulmonar destacan unos crepitantes secos inspiratorios bibasales, caquexia marcada y presenta artrosis de manos con nódulos de Bouchard y Heberden, leve sinovitis sin dolor en 2º IFP de mano derecha y esclerodactilia.
Ingresa en planta de Neumología con sospecha de infección respiratoria recibiendo tratamiento con antibioterapia, corticoides sistémicos, broncodilatadores y oxigenoterapia en gafas nasales con disminución progresiva de las necesidades de oxígeno.
En las pruebas complementarias realizadas destaca en la analítica sanguínea una proteína C reactiva de 84.2 mg/l y leucocitosis con desviación izquierda. En la radiografía de tórax PA y lateral (ver imagen 1 y 2) se aprecia una afectación reticular bilateral en probable relación con fibrosis pulmonar y se identifican imágenes pseudonodulares bilaterales que no permite su filiadas mediante esta técnica de imagen. No se pueden realizar comparativas con estudios previos ya que no se dispone de ellos.
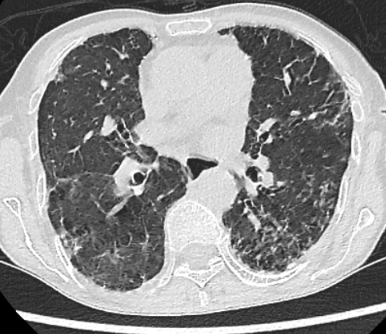
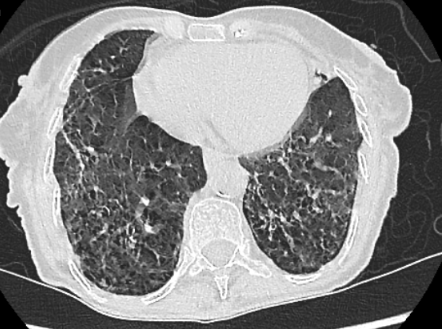
Se solicita TCAR donde se visualizan adenopatías supraclaviculares de hasta 7-8 mm de eje corto en lado derecho, indeterminadas. Se aprecia un calibre de la arteria pulmonar principal derecha en el límite alto de la normalidad y una marcada dilatación esofágica difusa hasta tercio distal sin cambios de calibre bruscos (ver imagen 3). Afectación difusa bilateral y heterogénea del intersticio pulmonar, con opacidades reticulares de predominio periférico y subpleural, con dudosa panalización en la periferia de los lóbulos inferiores (mal valorable debido a artefactos por movimientos respiratorios del paciente), con dudoso gradiente craneocaudal, identificándose bronquiectasias de tracción (ver imagen 4 y 5). No se observan claras consolidaciones ni nódulos de tamaño significativo. No hay derrame pleural ni pericárdico.
Con estos hallazgos se realiza una broncoscopia, fundamentalmente para descartar causa infecciosa y orientar el diagnóstico con el lavado broncoalveolar (LBA). La microbiología resulta negativa incluyendo las micobacterias y el LBA presenta un recuento celular normal.
En el estudio de autoinmunidad destaca ANA +1/1280 centromérico, anticentrómero positivo, antiCENP-A y B positivos, FR 22, anticitrulinados < 1.21. Resto de la autoinmunidad negativa.
La paciente presenta una evolución respiratoria favorable. Es valorada durante el ingreso por Reumatología y se procede al alta con tratamiento con corticoides sistémicos en pauta descendente manteniendo una dosis de prednisona de 10 mg/día hasta revisión en las consultas. En la revisión en consulta de enfermedades pulmonares intersticiales se reevalúa a la paciente con pruebas funcionales respiratorias que muestran un patrón respiratorio restrictivo y descenso moderado de la capacidad de difusión (FVC 1730 ml (56%), FEV1 1410 ml (60%), FEV1/FVC 81, TLC 2.89 (62%), DLCO 53%, KCO 82%). También se realiza un ecocardiograma que muestra signos de hipertensión pulmonar en grado moderado. Tras descartar infecciones latentes se decide iniciar tratamiento inmunosupresor con micofenolato de mofetilo con buena tolerancia y estabilidad clínica y funcional durante el primer año de seguimiento.
DISCUSIÓN-CONCLUSIONES
La evaluación de la enfermedad pulmonar intersticial (EPI) en pacientes con enfermedades del tejido conectivo (ETC) es un desafío debido a la heterogeneidad de la enfermedad, los diversos patrones radiológicos y los diferentes grados de gravedad que pueden manifestarse, dado que la enfermedad pulmonar, puede diagnosticarse en cualquier momento.
Para detectar la presencia de ETC ocultas, es imperativo llevar a cabo una evaluación exhaustiva y multidisciplinaria. La determinación de si la EPI está asociada a la ETC requiere exclusión de posibles etiologías alternativas y evaluación de las características clínicas. El abordaje multidisciplinario se considera el estándar para el diagnóstico y manejo de los pacientes con EPI, por lo que la participación de neumólogos, reumatólogos, radiólogos y anatomopatólogos en este equipo es fundamental.
La EPI y la hipertensión pulmonar (HTP) están asociadas a una morbilidad significativa, determinan el mal pronóstico en la esclerodermia y constituyen la principal causa de mortalidad en pacientes con ETC. En estadios iniciales los pacientes pueden estar asintomáticos, apareciendo la disnea en algún momento de la evolución de la enfermedad. El pronóstico parece estar determinado por la gravedad de las pruebas de función respiratoria (capacidad vital forzada, FVC) y la extensión de la fibrosis en la TCAR más que por el subgrupo histológico. Por ello, a pesar de la heterogeneidad, no se recomienda la realización de biopsia pulmonar ya que no suele cambiar el pronóstico ni el tratamiento. Sólo sería aconsejable si hay dudas de que la EPI sea debida a una ETC.
Se recomienda realizar pruebas funcionales respiratorias completas y radiografías de tórax en el momento del diagnóstico de ETC, así como un TCAR ante la mínima sospecha de EPID asociada. LA DLCO es el parámetro más sensible para detectar afectación pulmonar en las ETC. Es necesario realizar un ecocardiograma si se sospecha de hipertensión pulmonar.
Los marcadores serológicos relacionados con la ESP incluyen anticuerpos antitopoisomerasa (Scl-70), ARN polimerasa III, Th/To (con patrón de inmunofluorescencia nuclear) y anticuerpos anticentrómero (con patrón centromérico). La especificidad de estos anticuerpos tiene una alta capacidad predictiva de afectación pulmonar, observando que los anticuerpos antitopoisomerasa (Scl-70) y ARN polimerasa III se asocian a un elevado riesgo de desarrollar EPI y protegen frente al desarrollo de HTP aislada. En contraste, los anticuerpos anticentrómero, se relacionan fuertemente con el síndrome de CREST (calcinosis, fenómeno de Raynaud, dismotilidad de esófago, esclerodactilia y telangiectasias) y se asocian predominantemente con HTP y una baja prevalencia e incluso ausencia de EPID10,11.
Respecto al tratamiento mediante inmunosupresores para el control de la enfermedad de base, se recomienda en aquellos casos en los que exista un deterioro clínico y funcional. Si cumple los criterios de enfermedad fibrosante progresiva (empeoramiento de los síntomas, una disminución del 5-10% del valor absoluto de la capacidad vital forzada (FVC) y/o una disminución ≥ 10% en el valor absoluto de la DLCO durante un año de seguimiento, o un incremento de las imágenes radiológicas fibrosantes y/o aparición de imágenes que no existían previamente el único que ha demostrado reducir la caída del FVC es el nintedanib12.
En conclusión, la interacción entre la ESP y la EPID presenta desafíos clínicos significativos. Comprender los mecanismos subyacentes y avanzar en técnicas diagnósticas y terapias específicas son esenciales para mejorar la calidad de vida y pronóstico de estos pacientes.
BIBLIOGRAFÍA
- Solomon JJ. et al. European respiratory update: scleroderma lung disease. Eur Respir Rev 2013; 22:127.
- Fischer A, Lee JS, Cottin V. Interstitial lung disease evaluation: detecting connective tissue disease. Respiration 2015; 90:177-84.
- Capobianco J, Grimberg A, Thompson BM, Antunes VB, Jasinowodolinski D, Meirelles GS. Thoracic manifestations of collagen vascular diseases. Radiographics 2012; 32:33-50.
- Ysamat Marfá R, Benito Ysamat A, Espejo Pérez S, Blanco Negredo M, Roldán Molina R. La patología pulmonar asociada a las enfermedades del tejido conectivo. Radiología. 2013;55 (2):107-117.
- Morales-Cárdenas A, Pérez-Madrid C, Arias L, Ojeda P, Mahecha MP, Rojas-Villarraga A, et al. Pulmonary involvement in systemic sclerosis. Autoinmun Rev 2016; 15:1094-108.
- Vij R, Strek ME. Diagnosis and treatment of connective tissue disease-associated Interstitial Lung disease Chest 2013;143(3):814-24.
- Morales-Cárdenas A, Pérez-Madrid C, Arias L, Ojeda P, Mahecha MP, Rojas-Villarraga A, et al. Pulmonary involvement in systemic sclerosis. Autoinmun Rev 2016; 15:1094-108
- ATS/ERS. International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias Am J Respir Crit Care Med 2002;165: 277-304.
- Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A et al.SENSCIS Trial Investigators. Nintedanib por systemic sclerosis-associated intersticial lung disease. N Engl J Med.2019: 380 (26): 2518-28.
- Bahmer T, Romagnoli M, Girelli F, Claussen M, Rabe KF. The use of auto-antibody testing in the evaluation of interstitial lung disease (ILD) – A practical approach for the pulmonologist. Respiratory Medicine 2016; 113:80-92.
- Mathai SC, Danoff SK. Management of interstitial lung disease associated with connective tissue disease. BMJ 2016; 352:1-14.
- Flaherty KR, Wells AU, Cottin V et al. Nintedanib in progressive interstitial lung diseases: data from the whole INBUILD trial. Eur Respir J. 2022; 59(3): 2004538.
ANEXOS
IMAGEN 1: Radiografía de tórax PA.
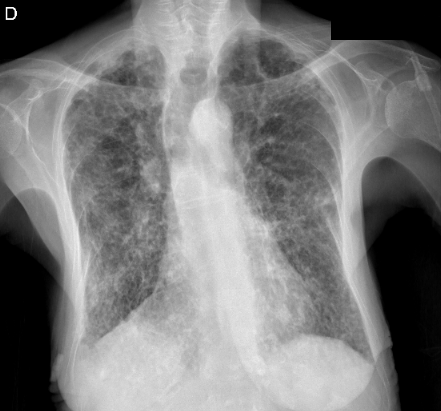
IMAGEN 2: Radiografía de tórax lateral.
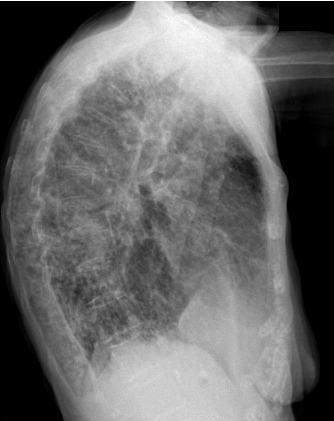
IMAGEN 3: TCAR corte transversal, ventana partes blandas.

IMAGENES 4 y 5: TCAR corte transversal, ventana pulmonar.