

AUTORES
- Nerea Frías Aznar. Médico de Atención Primaria en Centro de Salud Valdefierro, Sector III de Zaragoza.
- Pedro Isarre García de Jalón. Médico de Atención Primaria en Centro de Salud de Jaca, Huesca.
- Cristina Muniesa Urbistondo. Médico de Atención Primaria en Centro de Salud de Híjar, Teruel.
- Bárbara Pérez Moreno. FEA Servicio de Urgencias. Hospital Royo Villanova, Zaragoza.
- Alicia Pueyo Ucar. Médico de Atención Primaria en Centro de Salud de Aliaga, Teruel.
- Nuria Alegre Villarroya. Médico de Atención Primaria en Centro de Salud Monreal del Campo, Teruel.
RESUMEN
El penfigoide ampolloso es una enfermedad autoinmune caracterizada por lesiones ampollosas causada por anticuerpos dirigidos contra la membrana basal. Afecta en su mayoría a la población anciana y es considerada la enfermedad ampollosa más frecuente en los países desarrollados, con una incidencia de 0,2 a 3 casos nuevos por cada 100.000 habitantes en nuestro medio.
Hasta el momento, son varios los medicamentos que pueden inducir penfigoide ampolloso: antibióticos, antihipertensivos, diuréticos, vacunas, fármacos anti-TNF-α y los inhibidores de la dipeptidilpeptidasa-4 (iDPP-4 o gliptinas); estos últimos con evidencia sólida de poseer riesgo alto de favorecer la aparición de esta enfermedad.
Su retirada en pacientes que han desarrollado la enfermedad autoinmune mejora el pronóstico.
PALABRAS CLAVE
Penfigoide ampolloso, gliptinas.
ABSTRACT
Bullous pemphigoid is an autoinmune disease characterized by bullous lesions caused by antibodies directed against the basement membrane. It mostly affects the eldery population and is considered the most common blistering disease in developed countries, with an incidence of 0.2 to 3 new cases per 100,000 inhabitants in our setting.
To date, there are several medications that can induce bullous pemphigoid: antibiotics, antihypertensives, diuretics, vaccines, anti-TNF-α drugs and dipeptidylpeptidase-4 inhibitors (iDPP-4 or gliptins); the latter with solid evidence of presenting a high risk of promoting the appearance of this disease.
Withdrawal of these medications in patients who have developed the autoimmune disease improves the prognosis.
KEY WORDS
Bullous pemphigoid, gliptins.
INTRODUCCIÓN
El penfigoide ampolloso es una enfermedad cutánea autoinmune caracterizada por lesiones ampollosas mediada por autoanticuerpos dirigidos contra la membrana basal (antígenos proteicos BP230 y BP180). Afecta en su mayoría a la población anciana, pero puede afectar también a jóvenes y niños. Es considerada la enfermedad ampollosa más frecuente en los países desarrollados, con una incidencia de 0,2 a 3 casos nuevos por cada 100.000 habitantes en nuestro medio1-7.
La patogenia del penfigoide ampolloso está definida por un componente inmunológico (anticuerpos frente a proteínas de la membrana basal) y un componente inflamatorio (activación de neutrófilos y eosinófilos por la fracción Fc de los anticuerpos; responsables de dañar la unión dermo-epidérmica). Además, son varios los factores desencadenantes de la aparición del penfigoide ampolloso: enfermedades neurodegenerativas, fototerapia, radioterapia, vacunas, infecciones víricas así como fármacos (antibióticos, antihipertensivos, diuréticos, fármacos anti-TNF-α y los inhibidores de la dipeptidilpeptidasa-4)1-3,5,7.
La eliminación del factor desencadenante mejora el pronóstico y facilita el manejo de la enfermedad7.
PRESENTACIÓN DEL CASO CLÍNICO
Mujer de 95 años con antecedentes personales de hipertensión arterial, dislipemia, diabetes mellitus tipo II, fibrilación auricular, cardiopatía isquémica y enfermedad renal crónica. En tratamiento con Diltiazem, Simvastatina, Rivaroxaban, Sitagliptina, Nitroglicerina y Xipamida.
Desde hace cuatro meses presenta erupción vesículo-ampollosa en tronco y extremidades (ver Imágenes 1, 2 y 3). Inicialmente, las lesiones son pápulas de menos de un centímetro de diámetro, localizadas a nivel de escote y espalda que posteriormente se convirtieron en lesiones costrosas. Dada la clínica de la paciente, se plantearon los siguientes diagnósticos diferenciales: eccema de contacto, eritema multiforme, pénfigo y penfigoide.
Ante la presentación clínica de la paciente, se inicia tratamiento con clobetasol tópico y retirada de sitagliptina; con mejoría parcial. A pesar de ello, las lesiones fueron aumentando de tamaño, extendiéndose a otras localizaciones; con erupción vesículo-ampollosa de contenido seroso en tronco, abdomen, tercio superior de espalda y extremidades con importante afectación palmoplantar, donde las lesiones son de contenido serohemorrágico.
Presenta pequeñas erosiones en mucosa oral e intertrigo a nivel inguinal bilateral, axilar derecho y pliegues abdominales, así como eritema y erosiones en área genital, por lo que requirió ingreso hospitalario.
Durante el ingreso hospitalario la paciente recibe tratamiento sistémico con metilprednisolona, prednisona, amoxicilina-clavulánico, antihistamínicos y analgésicos. Además, se realizaron curas tópicas de las lesiones. Se realiza biopsia, el informe describe una lesión ampollosa subepidérmica con contenido de eosinófilos, compatible con penfigoide ampolloso. Se observa depósito en membrana basal de C3 e IgG. El resto de los anticuerpos estudiados han resultado negativos (IgA, IgM, C4c, C1q, fibrinógeno) y en la inmunofluorescencia indirecta se observa positividad de los anticuerpos contra la membrana basal epitelial (+1/80) y negatividad de los anticuerpos contra la sustancia intercelular.
Tras 15 días de ingreso hospitalario las lesiones cutáneas evolucionaron favorablemente (ver Imágenes 4, 5 y 6), por lo que se decide revisión en consultas externas. Actualmente, la paciente se encuentra asintomática y con reepitelización de las lesiones.
DISCUSIÓN
El penfigoide ampolloso se caracteriza por lesiones muy pruriginosas de aspecto urticariforme o eccematosas en estadíos iniciales sobre las que aparecen ampollas tensas con contenido seroso o hemorrágico. Se debe evitar el infradiagnóstico en ancianos ya que en ocasiones cursa con un cuadro de prurito crónico. Su localización afecta al tronco y flexuras, respetando la cabeza y el cuello1-3.
El diagnóstico se basa en la historia clínica, así como en los datos inmunológicos e histológicos. Además, la presencia de tres de los siguientes cuatro criterios: ausencia de afectación en mucosas, ausencia en cabeza y cuello, ausencia de cicatrices atróficas y la afectación en pacientes mayores de 70 años presentarían una especificidad del 83%, una sensibilidad del 90% y un valor predictivo positivo de 95%1,5.
La confirmación diagnóstica se realiza con el estudio histológico tras la toma de muestra de una de las vesículas de reciente aparición; observando una ampolla subepidérmica con infiltrado inflamatorio perivascular superficial con abundantes eosinófilos1.
Para el estudio de la inmunofluorescencia directa, se debe tomar muestra de la zona perilesional; donde se observan depósitos lineales de IgG y C3 en la membrana basal4.
La proteína DPP4 (CD26) se expresa en células como los linfocitos T y aumenta en las enfermedades dermatológicas. En el caso que nos ocupa, el uso de un iDPP4 parece ser el factor desencadenante de la aparición de la enfermedad, ya que la inhibición de la formación de plasmina por DPP4 puede modificar la respuesta inmune (afectando a la actividad de proteasas con la degradación inapropiada del antígeno BP180, alterando propiedades antigénicas de la membrana basal, mejorando el reclutamiento dérmico de eosinófilos y la formación de ampollas); ya que el tratamiento con este tipo de antidiabéticos orales induce la generación de autoanticuerpos frente a las mismas proteínas. Su presentación clínica es similar a la presentación clásica con afectación de mucosas en el 30% de los casos. El inicio de los síntomas tras el inicio de tratamiento es de 7 meses de mediana2,4-7.
La incidencia estimada de penfigoide ampolloso en pacientes con diagnóstico de diabetes y tratamiento con iDPP-4 es de 0,86 por cada 1.000 habitantes4.
Es importante conocer el diagnóstico diferencial de las enfermedades ampollosas, diferenciando el grupo de los pénfigos; que cursa con ampollas frágiles, intradérmicas con signo de Nikolsky positivo del grupo de los penfigoides, que cursan con ampollas subepidérmicas como la dermatitis herpetiforme, la dermatosis IgA lineal, la epidermólisis ampollosa adquirida y el penfigoide en mucosas1.
El tratamiento médico consiste en retirar el iDPP-4, el uso de corticoides tópicos como antiinflamatorios, corticoides sistémicos como frenadores de la producción de anticuerpos patógenos o inmunoglobulinas endovenosas o plasmaféresis que favorezcan la eliminación de dichos anticuerpos1,3,4,6,7.
La mayoría de las lesiones mejora tras el mes de tratamiento, pero el pronóstico es variable, ya que depende de la supresión de la enfermedad al no tener un tratamiento curativo pero la mortalidad ha mejorado gracias al uso de la corticoterapia. En general, suele ser una enfermedad autolimitada pero la afectación en personas de edad avanzada, frágiles, con comorbilidades presenta mayor riesgo de complicaciones1,6,7.
CONCLUSIONES
El penfigoide ampolloso es una enfermedad dermatológica de causa autoinmune. Se debe a una producción de anticuerpos frente a la membrana basal. Su diagnóstico es clínico, inmunológico e histológico y se ha observado fundamentalmente en la población mayor de 75 años. Además, existen muchos fármacos que favorecen la aparición de la enfermedad; entre ellos, los iDDP4, cuya rápida retirada supone una mejora progresiva de las lesiones. El tratamiento se basa en corticoterapia tópica o sistémica, con antibióticos en caso de sobreinfección, el uso de inmunoglobulinas y plasmaféresis en casos más severos; así como en la retirada del fármaco1,3,5-7.
BIBLIOGRAFÍA
- Fuertes de Vega I, Iranzo-Fernández P, Mascaró-Galy JM. Penfigoide ampolloso: guía de manejo práctico. Actas Dermosifiliogr. 2014;105:328–346.
- Tasanen K, Varpuluoma O, Wataru N. Penfigoide ampolloso asociado al inhibidor de la dipeptidil peptidasa-4. Inmunol frontal. 2019: 10:1238.
- Pérez-López I, Moyano-Bueno D, Ruiz-Villaverde. Penfigoide ampolloso y vacuna COVID-19. Medicina Clínica. 2021; 157: e333–e334.
- Casasnovas AG. Penfigoide ampolloso asociado a inhibidores de la dipeptidil peptidasa 4. Aten Primaria. 2023; 55(4): 102587.
- Verheyden MJ, Bilgic A, Murrell DF. A systematic review of drug-associated bullous pemphigoid. Acta Derm Venereol 2020; 100.
- Martín-Enguix D, Ruiz-Villaverde R, Galán-Gutiérrez M, Cabrerizo-Carvajal AM. Asociación entre penfigoide ampolloso y los inhibidores de la dipeptidil peptidasa-4, e impacto de su retirada. Aten Primaria. 2023; 55(4): 102586.
- Magdaleno-Tapial J, Valenzuela-Oñate C, Esteban Hurtado A, Ortiz-Salvador J.M., Subiabre-Ferrer D et al. Asociación entre penfigoide ampolloso e inhibidores de la dipeptidilpeptidasa- 4: estudio de cohortes retrospectivo. Actas Dermosifiliogr. 2020;111(3):249-253.
Imagen 1. Afectación inicial con ampollas tensas en pie de contenido seroso y lesiones costrosas.
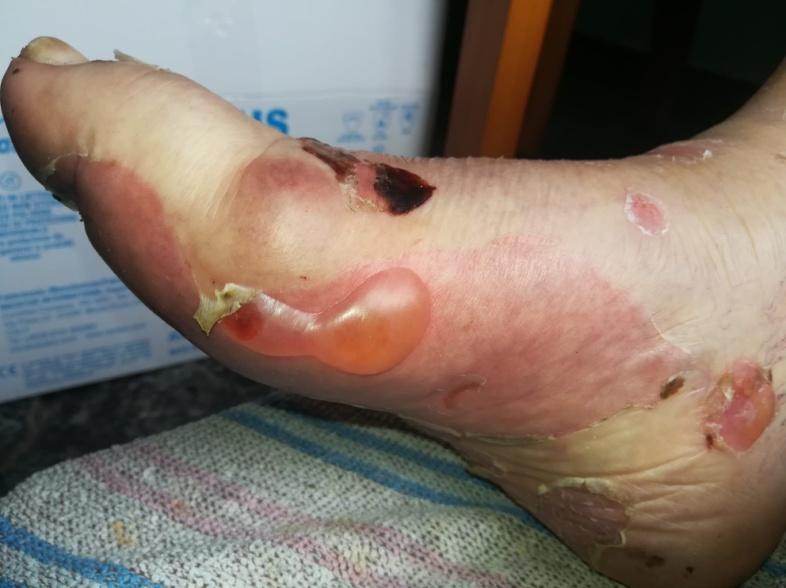
Imagen 2. Afectación inicial con ampollas y lesiones costrosas en extremidades superiores.
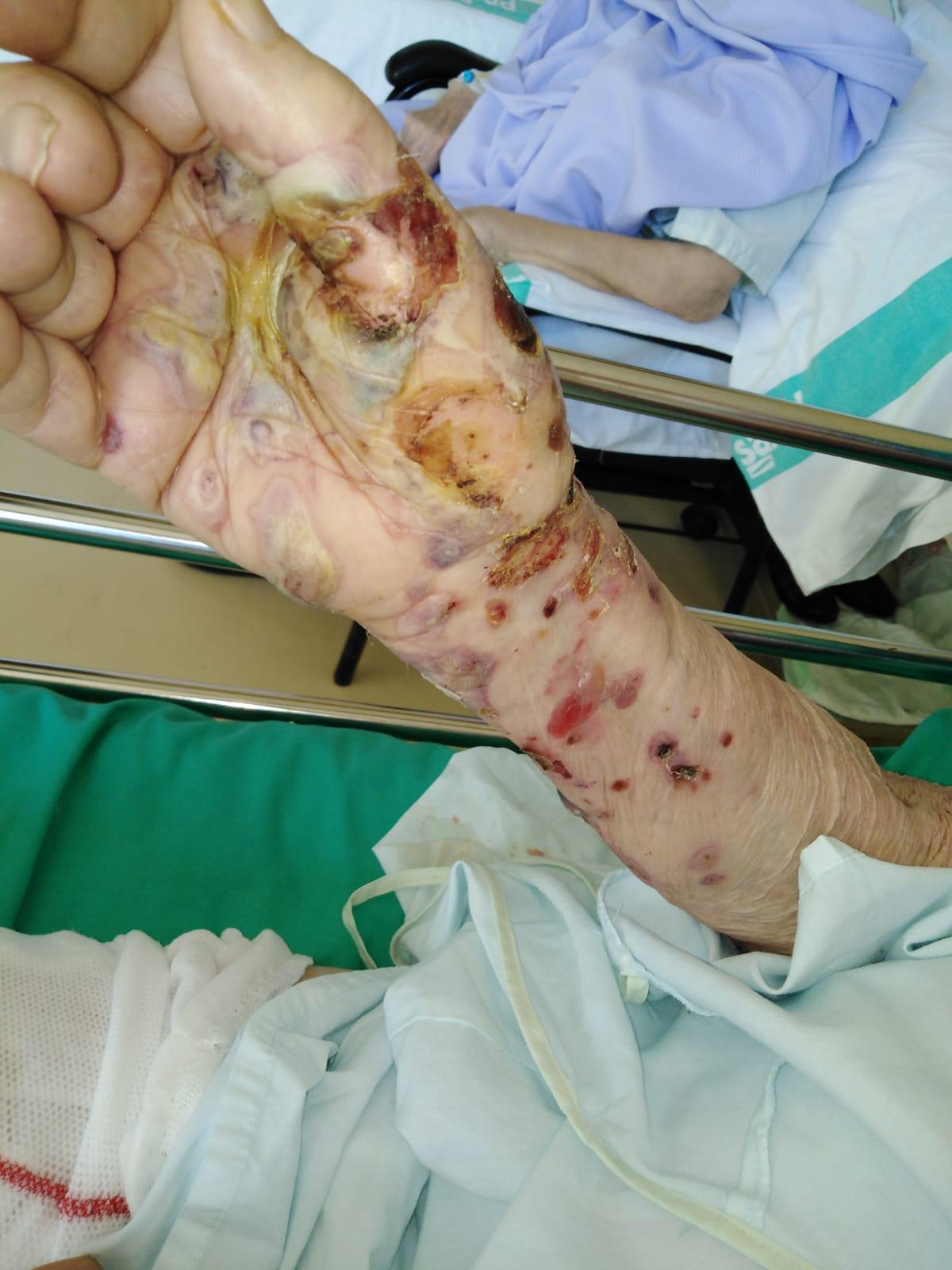
Imagen 3. Afectación inicial con ampollas en extremidades inferiores.
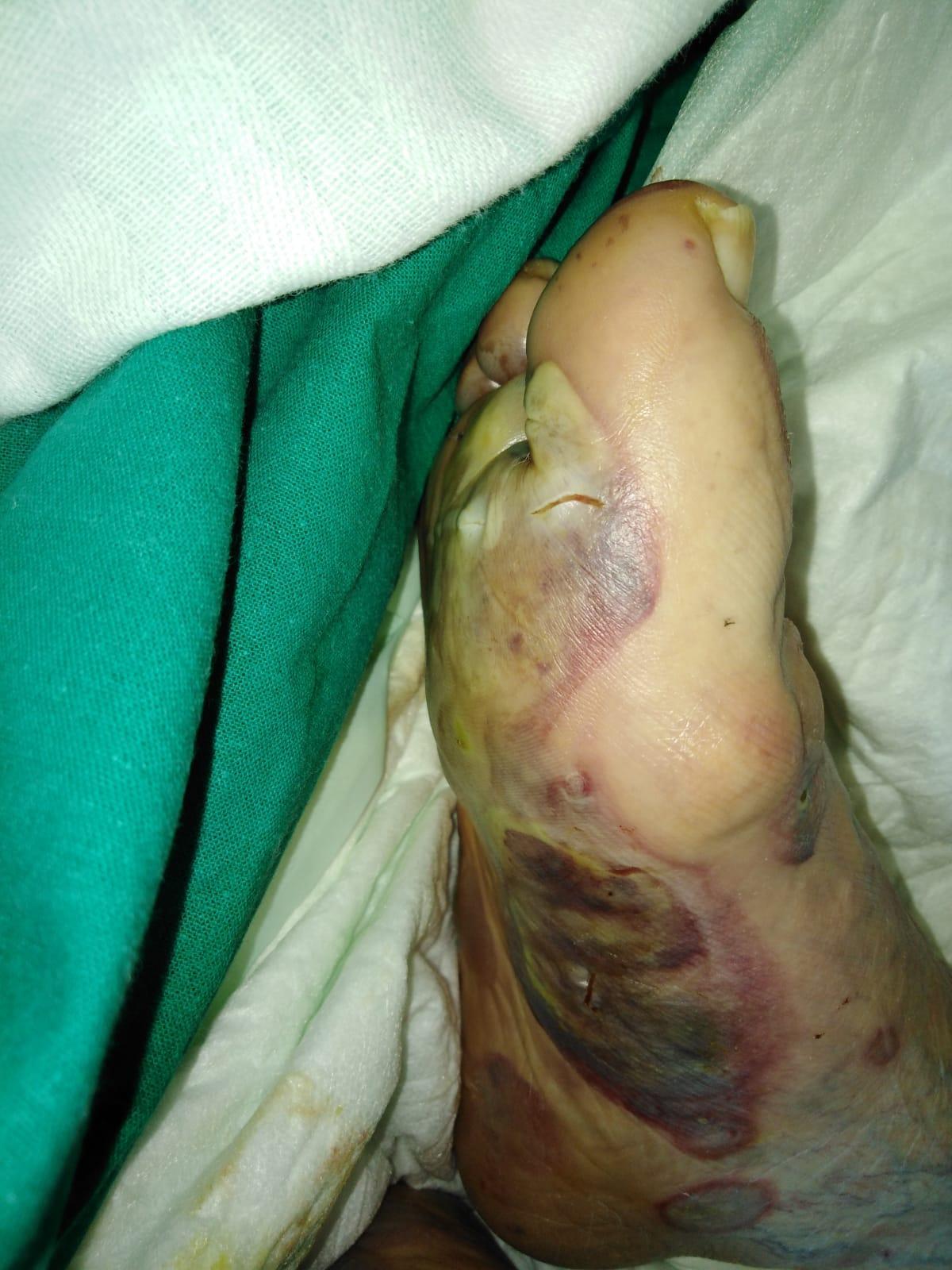
Imagen 4. Evolución tras tratamiento (extremidades superiores).
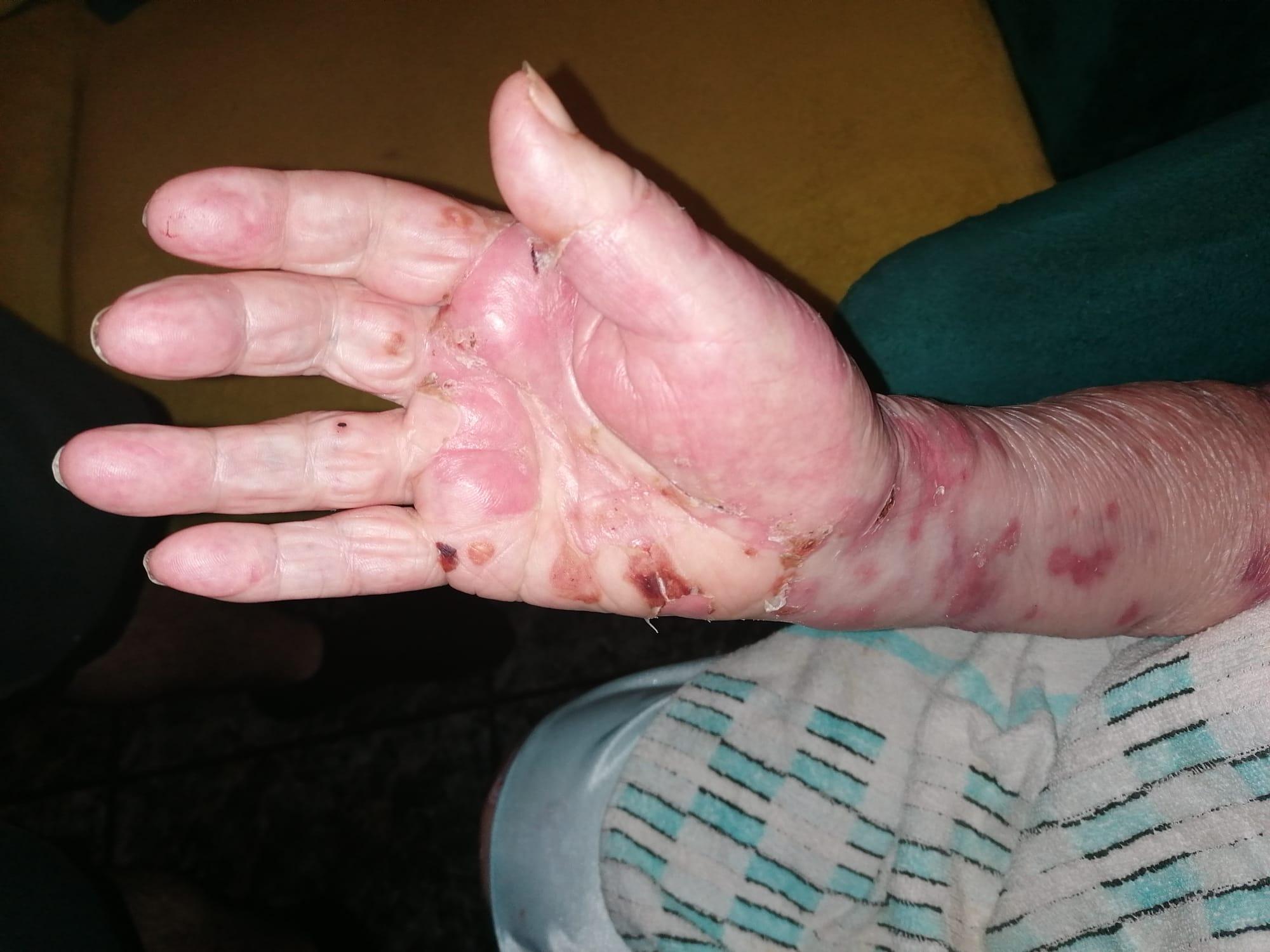
Imagen 5. Evolución tras tratamiento (dorso de extremidades inferiores).
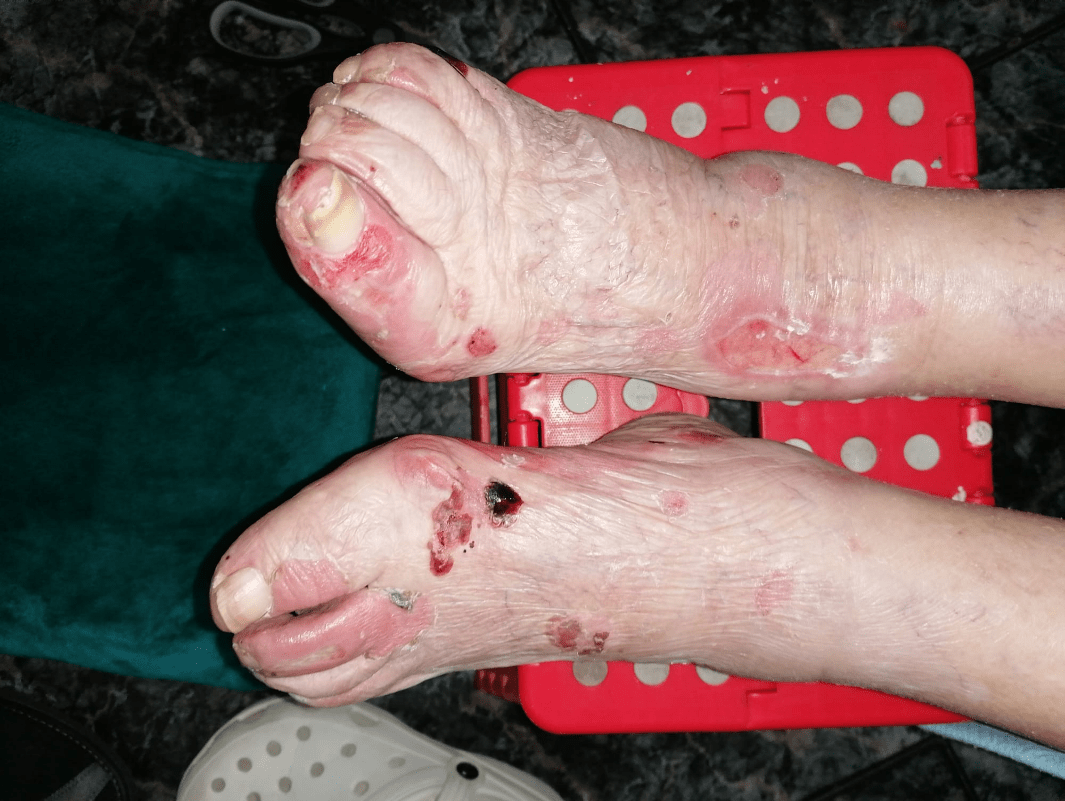
Imagen 6. Evolución tras tratamiento (planta de extremidades inferiores).
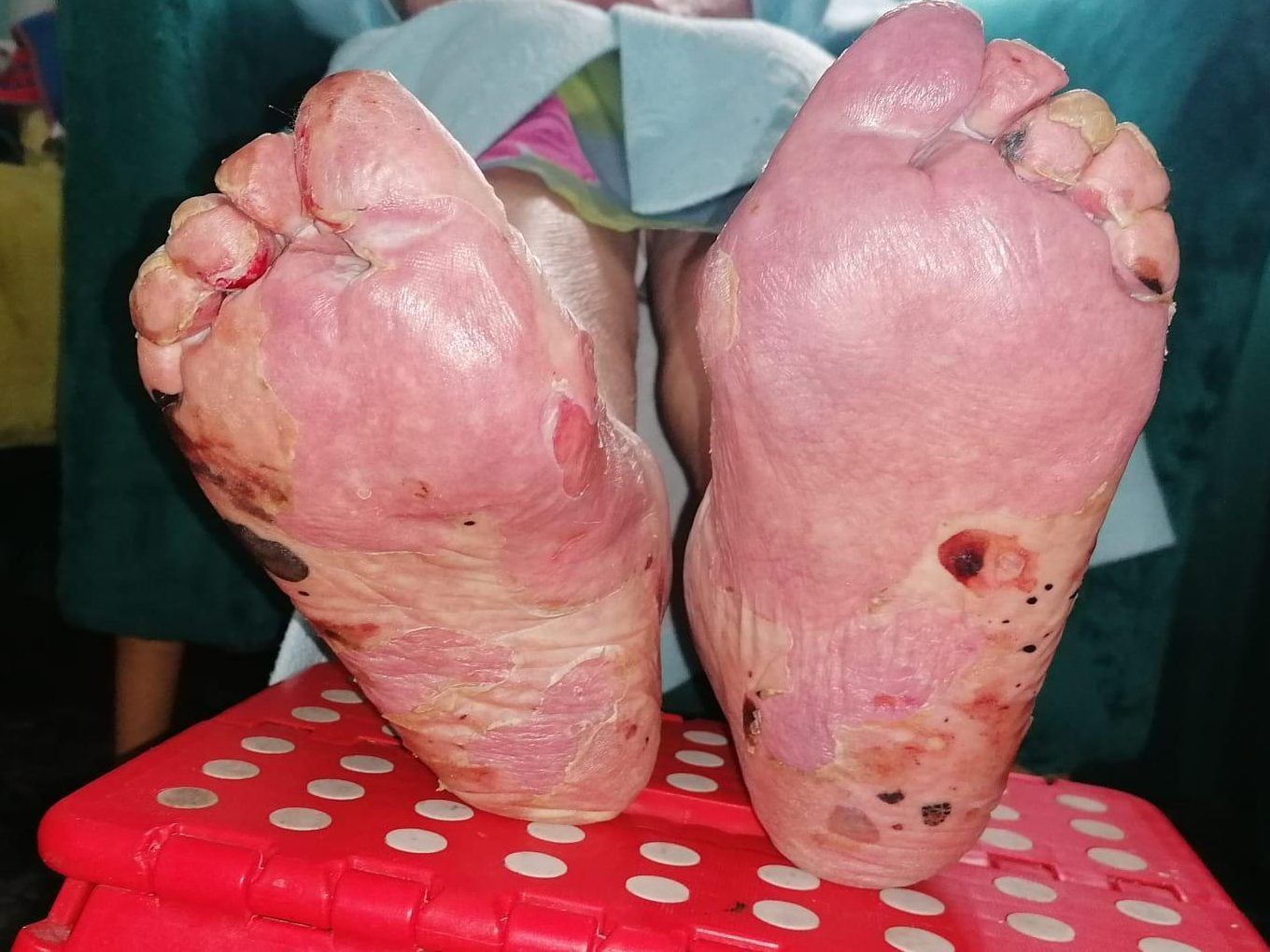
Todas las imágenes presentadas, son fotografías de elaboración propia tras consentimiento de la paciente.


